
中心体在G2/M期必须完成成熟,才能激活微管组织中心功能,确保双极纺锤体正确组装和染色体精准分离。PLK1是中心体成熟的关键激酶,其在中心体的时空定位直接决定成熟时机。然而,细胞周期如何调控PLK1的及时募集,此前尚不明确。近期发表于Nature Communications的一项研究,首次揭示了泛素E3连接酶RNF40通过“乙酰化-磷酸化转变”与CDK1-PLK1形成信号级联,从而实现中心体成熟的时间窗精准把控。该发现不仅阐明了中心体成熟的分子开关机制,也为肿瘤染色体不稳定性和化疗增敏提供了新靶点。
研究者首先通过免疫沉淀-质谱分析,在有丝分裂HEK293T细胞中筛选RNF40的相互作用蛋白,发现PLK1是显著富集的结合伙伴。内源及外源免疫共沉淀、GST pull-down实验进一步证实,RNF40的D4片段(222-628 aa)与PLK1的Polo-box结构域(PBD)直接结合。这表明RNF40可作为PLK1的潜在募集支架。
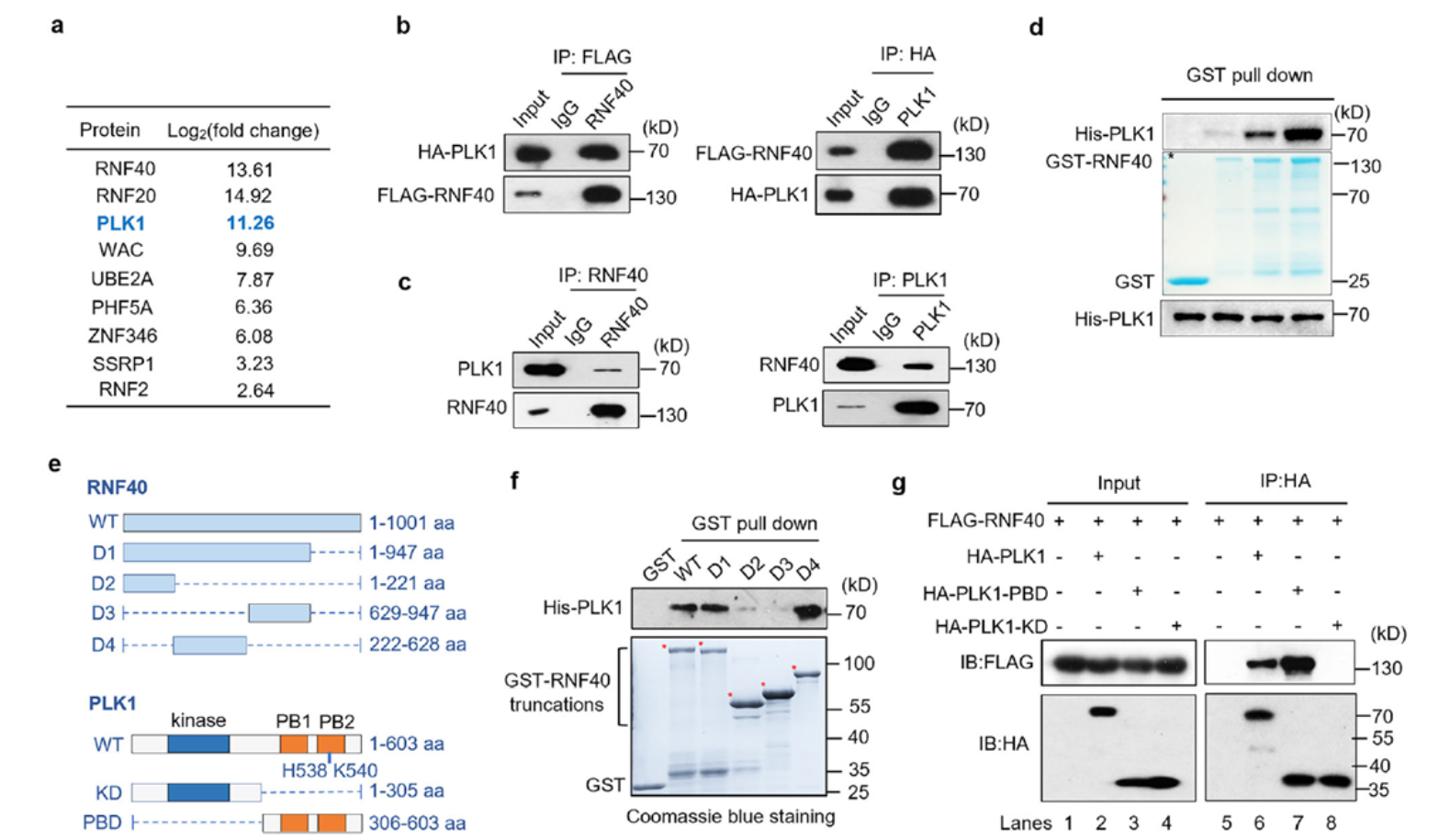
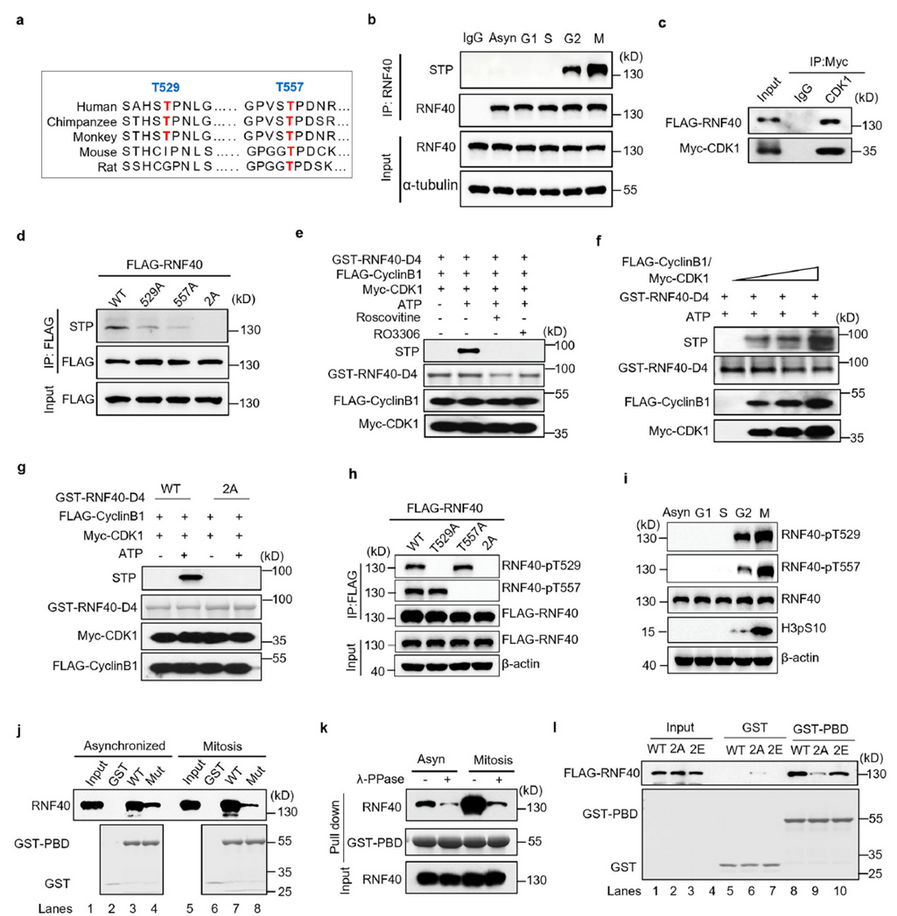
序列分析显示RNF40 D4片段含有两个保守的CDK1磷酸化位点(T529和T557)。细胞周期同步实验结合磷酸化特异性抗体检测表明,RNF40在G2期开始磷酸化,M期进一步增强。体外激酶实验证实CDK1/cyclin复合物可直接磷酸化GST-RNF40-D4片段,T529A/T557A双突变(2A)完全阻断磷酸化。
磷酸化直接促进RNF40与PLK1的结合,λ磷酸酶处理或CDK1抑制剂可削弱二者相互作用;2A突变体结合能力显著下降,而磷酸化模拟突变体(2E)维持强结合。由此确立了CDK1-RNF40-PLK1信号级联。至此,CDK1-RNF40-PLK1级联的磷酸化调控环节被清晰建立。
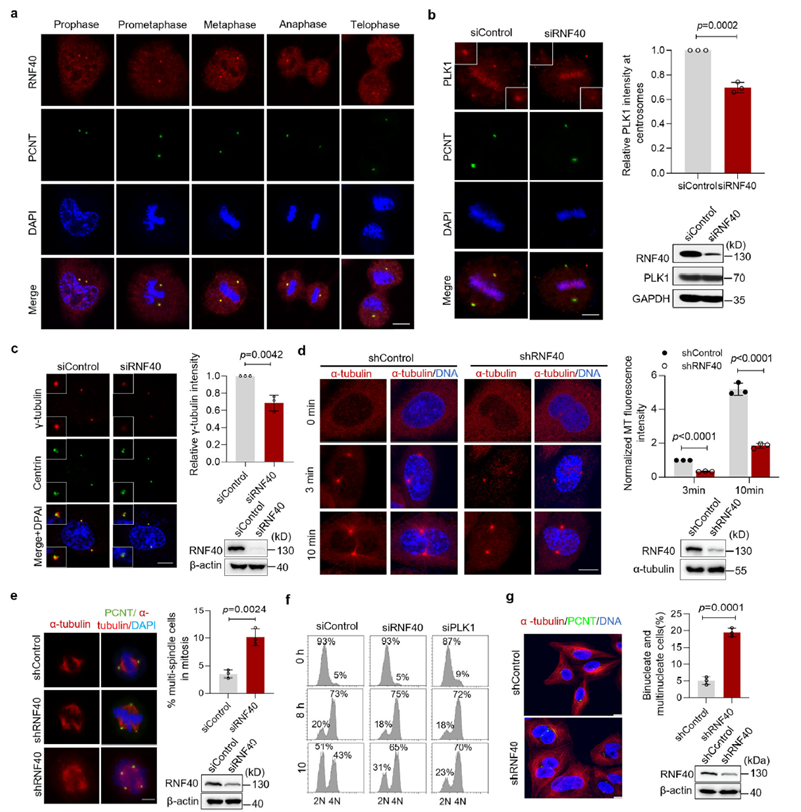
研究证实,RNF40在整个细胞周期中均定位于中心体,其定位依赖于CEP192/PCNT支架蛋白。功能实验显示,缺失RNF40会导致中心体处PLK1的募集显著不足,进而抑制γ-tubulin的积累与微管成核能力。这种功能受损最终会引发多极纺锤体组装、染色体排布异常及有丝分裂进程延迟。上述结果表明,RNF40通过介导PLK1的准确定位,成为驱动中心体成熟的关键正向调控因子。
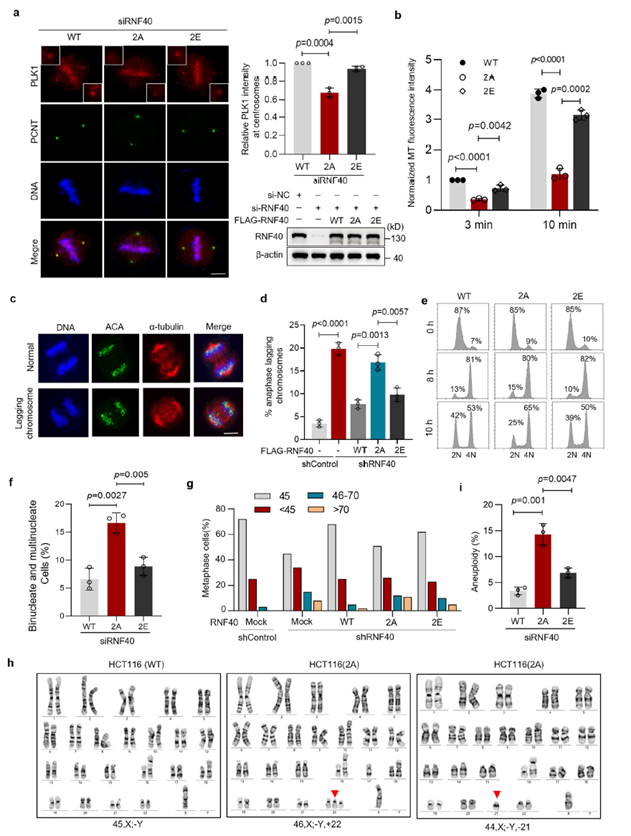
为验证磷酸化修饰的具体功能,研究者在缺失RNF40的细胞中分别回补了野生型、非磷酸化突变体(2A)及磷酸化模拟突变体(2E)。结果显示,2A突变体无法维持PLK1在中心体的定位,导致微管成核受损、染色体排列异常,并引发有丝分裂退出延迟及多核细胞增多;而2E突变体则能完全恢复这些功能缺陷。进一步分析证实,磷酸化缺失(2A)会直接导致染色体数目异常及基因组不稳定。值得注意的是,RNF40的这一调控功能完全依赖于其磷酸化状态,而非其传统的E3泛素连接酶活性。
为了探究RNF40在间期是否受到某种修饰的抑制,研究者发现RNF40在间期(G1/S期)会被PCAF乙酰化,而随着细胞周期进入G2/M期,该修饰又被HDAC1移除。实验表明,RNF40的乙酰化状态与其磷酸化存在明显的拮抗关系:若抑制去乙酰化过程,则RNF40的磷酸化水平及与CDK1的结合能力将显著下降。由此证明,RNF40存在“间期乙酰化锁定,G2/M去乙酰化并磷酸化激活”的动态转变。这一修饰状态的切换充当了信号传导的时间开关,精准调控着CDK1-RNF40-PLK1信号级联的激活时机。
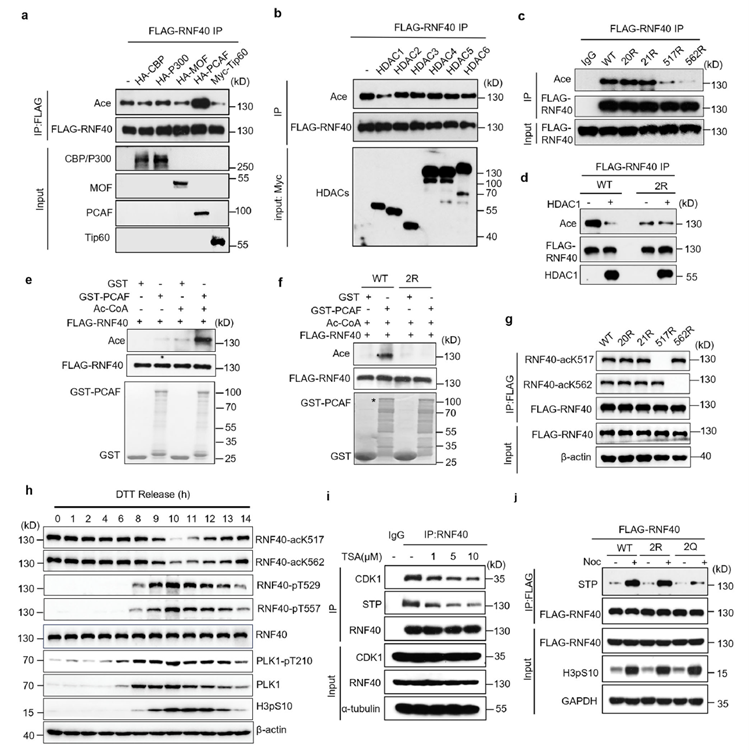
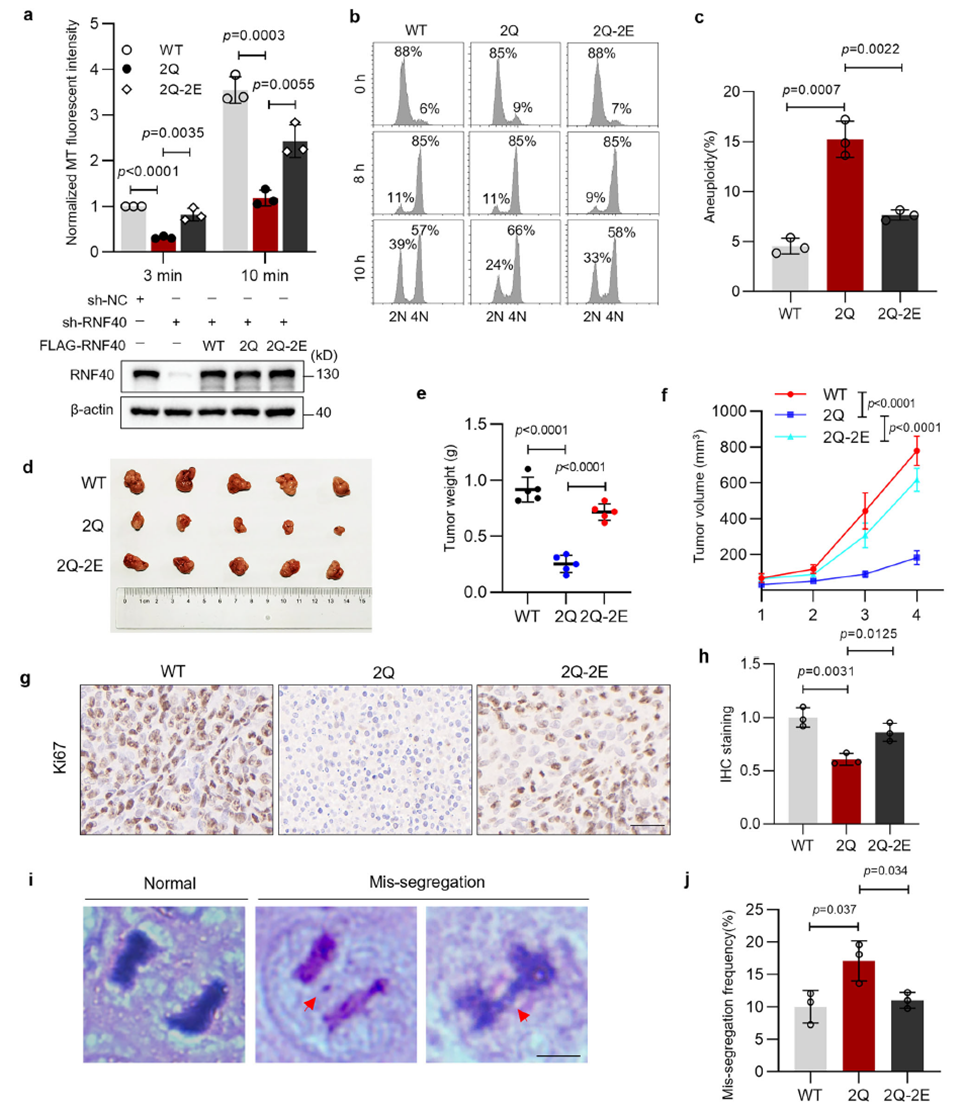
通过功能验证实验,研究者证实了RNF40“乙酰化-磷酸化”转变的生物学意义:持续的乙酰化修饰(2Q)会直接导致微管成核缺陷和非整倍体增加,而通过模拟磷酸化(2Q-2E)可完全逆转这一表型,证明其缺陷本质源于磷酸化水平不足。在动物实验中,表达乙酰化模拟突变体(2Q)的肿瘤生长显著受抑,表现为肿瘤体积缩小、增殖指数降低及染色体错分离增加。临床数据进一步支持了这一发现,RNF40在结直肠癌中呈高表达态势,且与癌症分期及淋巴结转移正相关。此外,RNF40发生的特定癌相关突变会通过阻断磷酸化修饰,诱发中心体功能缺陷。
该研究首次将RNF40定位为中心体成熟的时间节点,揭示了翻译后修饰动态转变在细胞周期中的精确调控策略,为理解有丝分裂保真性和肿瘤发生提供了全新视角,也为开发针对中心体-纺锤体异常的抗癌策略打开了新窗口。
AtaGenix技术支持
————————
AtaGenix定制开发高特异性的RNF40磷酸化抗体(pT529;pT557;acK517;acK562),助力研究团队清晰地捕捉到了RNF40修饰状态随细胞周期波动的动态轨迹,为确立CDK1-RNF40-PLK1信号级联的时间窗提供了核心证据,为后续开发靶向翻译后修饰的抗癌策略奠定了坚实的实验基础。
AtaGenix深耕翻译后修饰抗体定制领域,专注于高特异性抗体开发
已成功支持多篇高影响力细胞周期与肿瘤机制等研究


返回顶部
 扫一扫关注我们
扫一扫关注我们